 Italiano
Italiano English
EnglishI craniofaringiomi sono tumori relativamente rari che originano dalla ghiandola ipofisaria o in prossimità di essa.
I craniofaringiomi rappresentano dal 2.4 al 4 percento di tutti i tumori cerebrali, e sono più frequenti nei bambini che negli adulti.
Nonostante si tratti di tumori benigni, possono crescere rapidamente e hanno tendenza a recidivare dopo trattamento.
I tumori non sono solitamente scoperti finché non intaccano direttamente le strutture importanti circostanti, perciò spesso hanno dimensioni considerevoli al momento della diagnosi.
Cause
Non è chiara l’eziologia di questi tumori, ma si pensa originino dalle cellule residue di una struttura embrionaria chiamata tasca di Rathke.
Sintomi
I sintomi del craniofaringioma sono causati dalla compressione delle strutture adiacenti quando il tumore cresce.
Queste neoplasie sono solitamente localizzate vicino a strutture critiche, inclusa la ghiandola ipofisaria, l’ipotalamo, i nervi ottici e arterie importanti.
Se il tumore comprime il peduncolo ipofisario o coinvolge la ghiandola ipofisaria stessa, ad esempio, può determinare un deficit ormonale che può a sua volta condurre a
- un ritardo di crescita
- obesità
- altri problemi ormonali
Allo stesso modo, possono comparire dei deficit visivi qualora il tumore comprima i nervi ottici o le vie ottiche.
Neoplasie di grandi dimensioni possono bloccare il flusso del liquido cefalorachidiano, che bagna il cervello e il midollo spinale, causando idrocefalo.
Un aumento della pressione intracranica causa
- mal di testa
- nausea
- vomito
Diagnosi
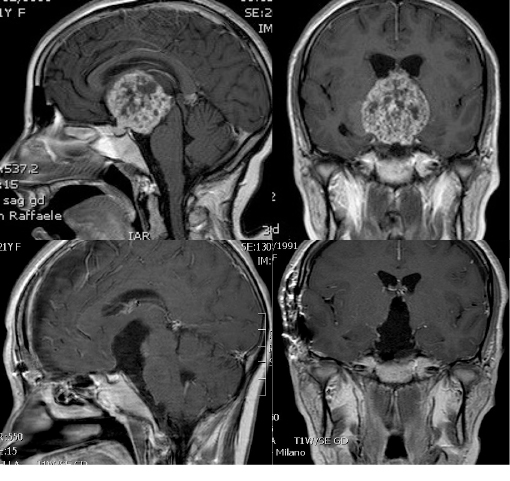
Gli studi di neuroradiologici sono la componente chiave per la diagnosi dei craniofaringiomi. Il mezzo di contrasto viene somministrato per via endovenosa così da meglio visualizzare il tumore rispetto al parenchima cerebrale di sfondo.
- Le immagini di Risonanza Magnetica (RM) sono importanti per determinare la relazione tra tumore e strutture adiacenti
- Anche la Tomografia Computerizzata (TC) è di aiuto
- In alcuni casi il neurochirurgo può utilizzare una RM con guida stereotassica “frameless”. Per questo studio, una RM con mezzo di contrasto viene acquisita dopo l’applicazione di marcatori di posizione speciali (chiamati fiduciali) alla cute del paziente. I fiduciali e le immagini neuroradiologiche sono processati mediante un software che calcola la localizzazione della neoplasia e crea una ricostruzione tridimensionale. L’immagine è quindi utilizzata durante la chirurgia per aiutare a localizzare precisamente la neoplasia, massimizzare la rimozione del tumore e minimizzare i danni al parenchima cerebrale circostante.
- Nei bambini affetti da craniofaringioma, è opportuno eseguire, inoltre, una valutazione endocrinologica e oftalmologica.
Trattamento
Il trattamento iniziale del craniofaringioma è abitualmente la rimozione chirurgica più radicale possibile cercando di preservare intatte le funzioni ipofisarie e neurologiche.
Purtroppo, a causa delle aderenze alle strutture adiacenti, a volte, il neurochirurgo si trova costretto a scegliere tra la rimozione totale, con un rischio di successivi problemi neurologici ed endocrinologici, e una resezione incompleta, che non garantisce una sicurezza sulla ricrescita tumorale.
Anche nei casi di rimozione totale questi tumori tendono a recidivare, per cui successivamente al trattamento andranno condotti degli studi di Follow-up con RM e TC.
In alcuni casi, qualora siano presenti dei deficit ipofisari, sarà necessaria una terapia ormonale sostitutiva sul lungo termine.
La radioterapia è utilizzata come secondo passo dopo la chirurgia, specialmente nei casi in cui la resezione non sia stata radicale. Questo tipo di trattamento però potrebbe avere degli effetti secondari sullo sviluppo dei bambini. Inoltre, alcuni casi possono essere trattati con la radiochirurgia stereotassica, che implica l’utilizzo di un fascio di radiazioni altamente focalizzate che convergono a trattare le massa tumorale risparmiando il parenchima cerebrale sano circostante.